
Material Science Group's Newsletter-2020
Editorial Board
Prof. Dr. Abdul Majid (Group Leader)
Ms. Alia Jabeen (Editor)
Ms. Rushba Zulfiqar (Editor)
Contents
• Editorial
• About the group leader
• Research facilities
• Research Group
• Group discipline
• Future planning
• Publications
Editorial
The usage of first principles calculations has emerged as a dependable tool for predicting material properties and fostering a deeper understanding of scientific mechanisms. Despite the extensive experimental efforts dedicated to synthesize novel materials, there has been relatively less emphasis on conducting Density Functional Theory (DFT) studies of these materials. However during past years, the DFT approach has got enormous attention to the community of theorists as these calculations based study enable us to replicate the real-world experimental conditions, aiding in our comprehension of the complicated relationship between a material’s structure and its electronic properties. DFT studies are renowned for their ability to save valuable time, energy, and financial resources for experimental material scientists. Modern computational systems provide theoretical forecasts of materials that are well-studied for
various applications. Our researchers and scholars are making it possible to achieve their scientific goals through pursing skills in simulating soft wares. Even their passion was appreciable during the period of COVID times in 2020. During that time, amid the COVID-19 pandemic, when traditional in-person methods became impractical or unsafe, our research scholars continued their work while adhering to social distancing guidelines and lockdown measures and conducted research through online mode via remote access. The collaboration and calmness among students and supervisor through that period was commendable, as all the students successfully completed their thesis along with outstanding publications.
About the Group Leader
Prof. Dr. Abdul Majid established this research group in 2010, focusing on semiconductors and diluted magnetic semiconductors for spintronic applications. His inspiration came from his tenure as a senior research associate at the Institute of Semiconductor Physics in Beijing, China, where he conducted an extensive investigation into ion-implanted compound semiconductors. After this, during his post-doc, the work was conducted using Density Functional Theory (DFT) with Vienna ab-initio simulation packages (VASP).
Apart from this, he expanded his research into predicting new materials and published findings in prestigious international journals. Later he introduced new solutions to existing problems by predicting new materials for Li ion batteries electrode, gas sensing and catalytic process. He also successfully predicted new layered materials.
"Your positive actions combined with the positive thinking results in success"
Research Facilities
Most students in this research group are engaged in studying diverse energy materials by employing DFT theory on a range of software applications installed on core i-7 computers. One of the hallmarks of DFT is its reliance on powerful computational tools. We use specialized software packages to perform complex calculations that simulate the behavior
of electrons in atoms and molecules. These calculations provide insights into the electronic structure, energy, and properties of materials, making it possible to predict their behavior without conducting extensive physical experiments. However, those students interested in conducting experimental work are also encouraged to pursue such endeavors
Research Codes
ü ADF
ü ADF-BAND
ü Reaxx-FF
ü Molecular Dynamics (MD)
ü DFTB
ü COSMO-RS
ü UFF (Universal Force Field)
ü GUI (Graphical User Interface)
ü Mopac
ü MM
Their functionality and other details could be found at www.scm.com
Research Students
Ms. Alia Jabeen
Ms. Alia Jabeen has recently completed her MPhil from University of Gujrat under the very kind supervision of Dr. Abdul Majid Sandhu. During her MPhil research, she has worked on structural and electronic properties of the transition metal oxides and observing the trends the dimensionality changes. Currently, she is focusing on the two dimensional layered materials. Her research interest includes the theoretical study of the material sciences and also keen to learn the basic mechanism behind the famous phenomenon of physics and DFT Calculations,

Ms. Amber Batool
Ms. Amber Batool is theorist working on Topological insulators. She did her M.Sc in Physics from university of Gujrat while working to investigate the clusters. She attempted to improve the understanding of structural, vibrational and optical behavior of TiSiO4 hybrid cluster using DFT in Amsterdam density functional (ADF) code under the supervision of Dr. Abdul Majid Sandhu. Her research interests include computational study of material science with focus on semiconductors, energy materials and devices. She got 8th position in Photo contents “Science in Nature” at international level held by Gull’s Association. Currently while perusing M.Phil, she is focusing on 2D topological insulators that promise an avenue to realize fascinating application such as dissipation less transport, spintronic, optoelectronics, thermos-electronics and fault-tolerant quantum computing. She wants to develop more promising topological insulating material and investigate electronic, spin-momentum locked topological edge states and spin orbit coupling in 2D predicted topological insulators. Observation of Dirac cone in the band structure means a lot in spintronics applications

Ms. Ghazal Nadeem
Ms. Ghazal Nadeem is an MPhil research student studying in Department of physics, University of Gujrat, Pakistan. She had worked as an Assistant lecturer in same department. She did her BS in physics from the same department. The research paper named ” A DFT study of bismuthene as anode material for alkali-metal (Li/Na/K)-ion batteries has been published in the journal “Material Science and Engineering B”.
Ms. Sana Hamid
Ms. Sana Hamid Aziz is an MPhil research student, studying in Department of Physics, University of Gujrat, Pakistan. She has done her BS in physics from the same university. The research paper named “Computational Study of Borophene/Boron Nitride (B/BN) Interface as a Promising Gas Sensor for Industrial Affiliated Gasses” had been published in the journal” Physica E: Low-dimensional Systems and Nanostructures “.
Email: sananiqa12@gmail.com

Ms. Hajrah Kanwal
Ms. Hajrah Kanwal is currently
working to enhance the electrocatalytic and photo catalytic performance for
Hydrogen evolution reaction (HER) and oxygen evolution reaction (OER) during
water splitting to meet the energy demands. She did her BS in physics from
University of Gujrat.. Her BS research work was
to calculate and understand the thermal and structural properties of
2D-materials for making their use possible in high temperature device grade
applications. Several computational techniques as geometry optimization and molecular
dynamics were employed to relax the crystal structures and to study thermal
behavior in ReaxFF code under the supervision of Dr. Abdul Majid Sandhu. The
radial distribution function (RDF) and phase-transition played an important
role for the comparison study of most to least thermally stable 2D-materials.
Moreover, her research interests include energy storage (through rechargeable
batteries) and energy production (by improving the catalytic activity for HER)
by using novel 2D-materials. She used VASP
code for implementation of her research ideas.

Research Essay's
Description of Exchange correlation Functional for vdWs layered materials
by Alia Jabeen
The outcomes of the Kohn-sham and Hohenberg disclosed that ground state energy could be obtained by attaining the minimum energy of energy functional and it could be executed through the discovery of a self-consistent solution to a set of one particle equations. This could be attained only by solving the Kohn-sham equation, an exchange-correlation function and this might be done by explicitily writing their parts. Actually the exchange correlation functional notify about almost all types of quantum mechanical interaction in between the electronic densities. There are several functional that are utilized in the DFT such as local density approximations (LDA), generalized gradient approximations (GGA), meta-GGA, Hybrid Functional, dispersion functional. The choice of functional are based on the material and the accuracy of the functional is decided on the basis of Jacob’s Ladder.
In Local density approximation (LDA), the density of electron gas is constant throughout the environment and is applicable for the systems where the movement of the particles fluctuates steadily within the electron gas. However, LDA over binds the molecules and underestimates the band gaps of insulators as well as the semiconductors up to 40%. This underestimation of band gap is expected from unknown accurate/exact potential of the DFT due to the discontinuity of exchange correlation energy. The generalized gradient approximation (GGA) considers the gradient of the electron gas and calculates the electron density to a specific region. It also underestimates the band gaps but it predicts the energy trends well for various materials so taken into consideration. Several extensions of the GGA have been introduced that includes the Perdew-Burke Ernzerhof PBE, PBE-sol, PW-91 and so on. In contrast to LDA, this approximation is more precise and errors are remarkably decreased.
The solution of Kohn-Sham equation under LDA, considered as the appropriate method to compute the structural characteristics close to the equilibrium inter-layer distance of systems such as graphite as well as h-BN.
Also, the more detailed XC functional GGA fails drastically to describe the interlayer distancein graphene as well as in h-BN although a complete description of the covalent bonding is given by this method. The main drawback of the mostly utilized functional (i.e. LDA & GGA) is that it does not properly describe the non-local electronic correlation impacts e.g. van der Waals (vdWs) interactions. This is because of the absence of the elucidation of the vdWs interaction that are the demonstration of long range correlation impacts. For layered two dimensional materials that includes the strong in-pane covalent bonding and weak van der Waals interactions. The explanation of these systems must incorporate an exact investigation of the full range of interactions from the small range covalent bonding towards the long range vdWs forces.
Generally, the addition of the vdWs based DFT method could be categorized into two types. One includes the approach established on the semi-empirical amendments usually accompanied by the dispersion corrections of the Kohn sham energy and the other includes a non-local DFT that directly change the Hamiltonian of Kohn sham. The initiation of the dispersion methodology, entitled as DFT-D and DFT-D2 that utilizes the constant values of the coefficients “C6” for every chemical family. This estimation does not include the impact of several hybridization states or oxidation of the similar element. Hence, more advancement in coefficient C6 based on the environment of the chemical has been established to such methods. The DFT-D3 technique as proposed by the Grimme et al., involve the coefficients based on the environment by considering the number of adjacent atoms that each atom possess. A different technique as proposed by the Tkatchenko and Scheffler which established the C6 coefficients on new scale by considering reference atomic “C6” coefficients, reference atomic polarizability as well as the effectual atomic volumes attained by dividing the total density of electrons of the system b/w the single atoms utilizing the “Hirshfeld partitioning scheme”.
A DFT Study of Alkali metal adsorption on bismuthene as an anode material
by Ghazal Nadeem
In my research period, I decided to work on the energy storage devices which are at the heart of current
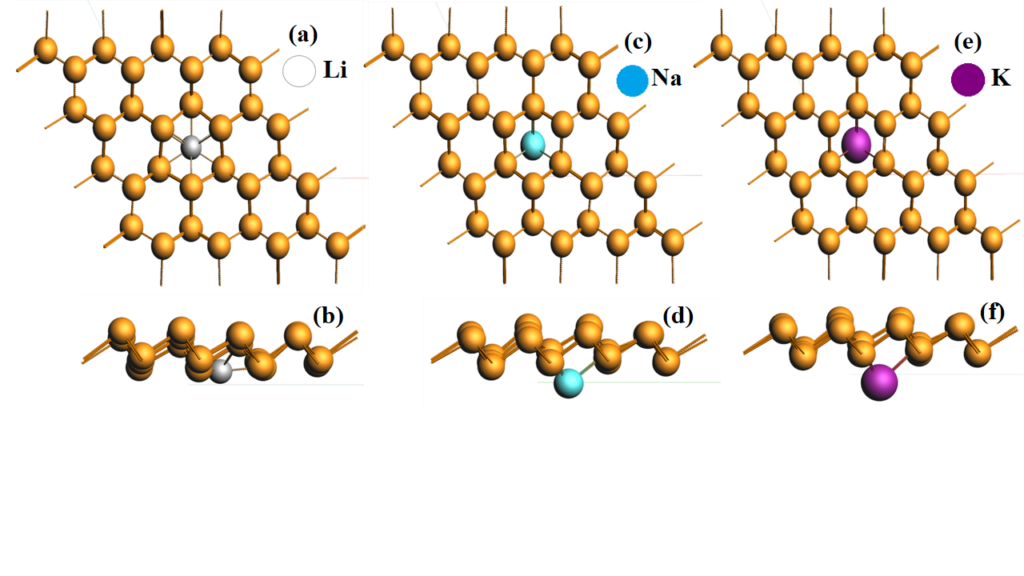
research activities for the future of humankind. I have chosen to work on the energy storage materials especially the materials involved in Lithium ion battery (LIBs), Sodium ion battery (NaIBs) and potassium ion battery (KIBs). Through the literature survey, I decided to work on bismuthene to finds its implementations as an anode material in LIBs, NaIBs and KIBs. The entire study is theoretical in nature using first principles calculations based on density functional theory (DFT). Firstly, I optimized its structure and studied its structural properties. Then I decided to adsorb the alkali metal (Li, Na and K) as shown in figure 1 on bismuthene in order to investigate its behavior after adsorption. Because of the semiconducting nature of bismuthene owes the greater stability to operate at or above room temperature. For the batteries to operate, it is necessary that the host material should show the metallic behavior. Gradually, increasing number of alkali- metal adsorption (Li, Na and K) on bismuthene replaces its semi
conducting behavior in favor of metallic one as sketched in Fig.1. In addition to study the electronic properties of the anode material, their migration properties are also estimated using climbing nudged elastic band (CI-NEB) simulations operating on density functional tight binding (DFTB) as sketched in the Fig. 2.
Prior to the previous studies, I have also calculated the specific theoretical capacities and open circuit voltage which are the important factors in the operation of batteries. My findings suggests that high storage capacity of 2275 mAh/g, 2149 mAh/g, 1896 mAh/g and extremely low diffusion barrier of 0.15eV, 0.06eV, 0.002eV for Li, Na and K respectively make bismuthene a potential candidate for lithium, sodium and potassium ion batteries. To conclude this, we have comprehensively explored a new 2D material bismuthene as a potential candidate to find its implementations as an anode material in LIBs, NaIBs and KIBs.
Computational studies of borophene/BN interphase as a promising gas sensor for industrial affiliated gases
by Sana Hamid Aziz
In our developing country, due to the increasing demand of technologies, the emissions of different toxic gases as CO, CO2, NO, NO2 and NH3 from industries, vehicles or any other sources are increasing day by day.
The emission of these gases in different concentration is a deliberate issue and it demands gas sensors. Adsorption of these gases are widely used for the sensing property. Two-dimensional (2D) materials create an extensive interest of researchers because of their novel electronic, optical, biocompatible, high charging capacity, large surface area, exclusive physical and chemical properties as compared to bulk materials. These characteristics are essentially important for different applications like electrodes and gas sensing. I have decided to work on the adsorption of different toxic gases in the gas sensing applications. In the group of 2D materials, borophene is a recent addition. . Borophene has metallic nature and low stability. To increase its stability, we made its interface with boron nitride that is pure insulator. Interface of borophene and boron nitride showed metallic behaviour and we utilized the properties of B/BN as a single material. Interface may increase the adsorption sites and improve adsorption energy that is good for gas sensing. Firstly, we select favourable adsorption sites with low formation energy and then adsorb different gases as CO, CO2, NO, NO2 and NH3 on the selected sites as shown in fig. 3. All the gases showed chemisorption and make bond with the host material while CO2 showed physiosorption behaviour as shown in figure.
To calculate the adsorption energy , the following formula was used:
E ads = Etot − (Einterface +Emol)
The study showed the high adsorption energies than previous work. In addition to the study, the structural and the electronic properties of interface before and after adsorption of gases is also investigated. Total density of states (DOS) of pristine borophene and interface with adsorbed gases is shown in fig. 4 below. Our results show metallic behavior in both cases for all gases as inferred from the absence of a band gap that is good for gas sensing. In case of interface, for all the gases, it can be seen that these adsorbed gases induce change in DOS (broaden the peak) particularly in the valence band as compare to pristine borophene.
Similarly, the transfer of charge between the atoms of gas and the interface and the transport properties of the selected gases for gas sensing applications is also calculated. To know the performance of the interface as a better sensor, non-equilibrium Green’s function (NEGF) method is done with computation for the properties related to transport. To conclude this, we have succefully made the interface of borophene and boron nitride to use its metallics properties as a single material in the gas sensing applications to avoid from the toxic gases that are hazardous.


Group Discipline
In
this research group, achieving success relies on practicing patience,
maintaining a consistent work ethic, and prioritizing ongoing study. All
students are expected to adhere to group discipline, which includes regularly
submitting reports in a prescribed format on a weekly, monthly and semester
basis to keep the group and supervisor informed about their activities. Group
meetings are a forum for students to showcase their work, engage in rigorous
discussions and collectively elevate the research’s quality. The majority of
students are
immersed in the realm of density functional theory (DFT), which necessitates a deep understanding of relevant knowledge. At the beginning of their research journey, students are assigned a set of tasks that are important for comprehending and elucidating their work. Students in DFT labs develop intricate models to describe and simulate a wide range of physical phenomena. These models are finely tuned to replicate real-world systems, allowing scientists to explore the behavior of materials under varying conditions, such as temperature, pressure and chemical compositions.
Future Planning
Taking into consideration the latest research trends and especially the energy problems, we are planning to work on the
following,
· Study of new materials
·
Rechargeable
batteries
·
2D Catalytic
material
· Spintronic devices